Research Overview
The structure of biomolecular complexes influences their function, known as a structure-to-function relationship. In the Bricker Lab, we study these relationships for various biomolecular complexes across a wide range of length- and time-scales. With a multi-scale computational toolset, we can probe electronic-level effects from quantum mechanics calculations, molecular and system-level effects from statistical mechanics (all-atom molecular dynamics simulations), and energy transfer mechanisms using rate-based models. As an example of multi-scale interactions in biology, molecular-scale structural and electronic interactions are responsible for the unique energy transfer properties found in biological light-harvesting systems. Captured energy from sunlight is transferred and harvested at remarkable efficiencies approaching unity in certain cases. This is due to the specific molecular organization of chromophores scaffolded by protein assemblies, yielding efficient short- and long-range energy transport. We probe these short- and long-range interactions of molecular assemblies using electronic structure theory and multi-scale modeling in order to learn from nature and apply these organizational concepts to synthetic energy transport systems. While proteins are utilized as scaffolds for chromophores in natural light-harvesting complexes, we are interested in a broad set of techniques called DNA nanotechnology to create synthetic biological scaffolded materials. DNA nanotechnology utilizes the unique properties of double-stranded DNA, including predictable folding based on sequence complementarity to force DNA sequences into predetermined scaffolded arrangements, and can be built into multi-dimensional scaffolds.
Machine-learned electronic structure of nucleic acids
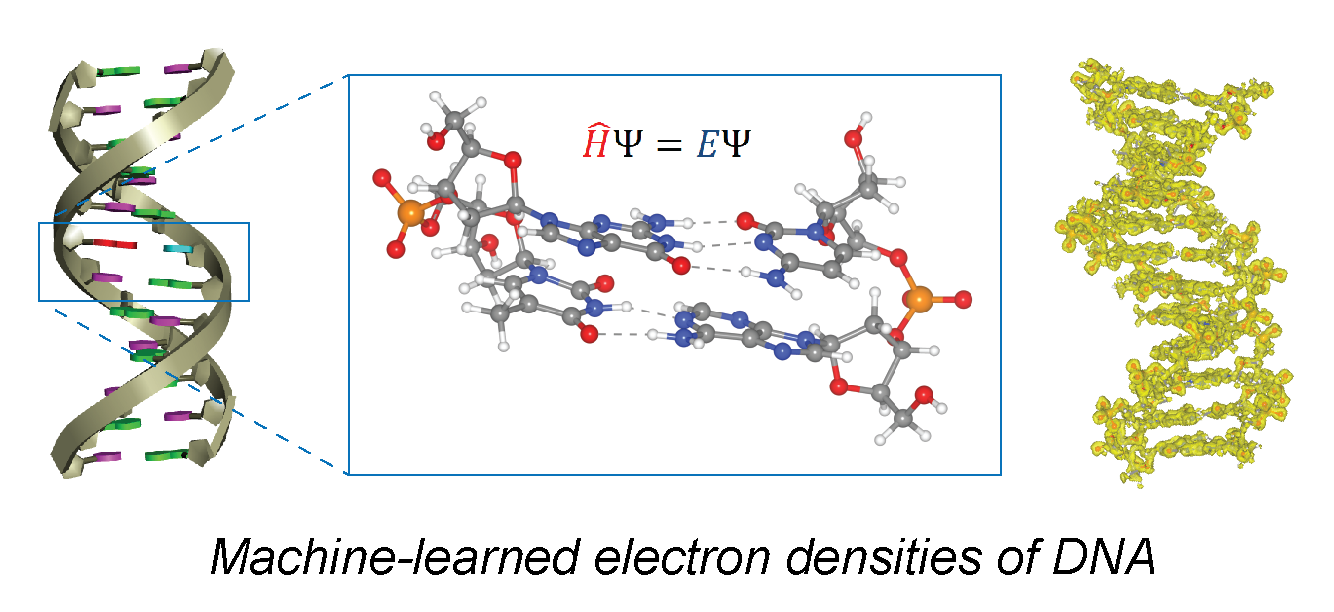
Accurate and efficient prediction of the electronic structure of biological systems such as nucleic acids could lead to breakthroughs in biophysical characterization, but using traditional quantum chemistry methods for these large systems are out of reach. With a structural ensemble from all-atom molecular dynamics, and ab initio calculations of relevant DNA fragments, we build a machine learning model using Euclidean neural networks to predict quantum-accurate electron densities for arbitrary DNA molecules. This ML model is accurate to within 1% error of the quantum reference calculations, scales linearly with respect to the number of atoms, and can include explicit solvation and ions by fragmenting the training set (Lee, Biophys. J., 2022; Lee, PLoS ONE, 2024).

Molecular aggregate structure
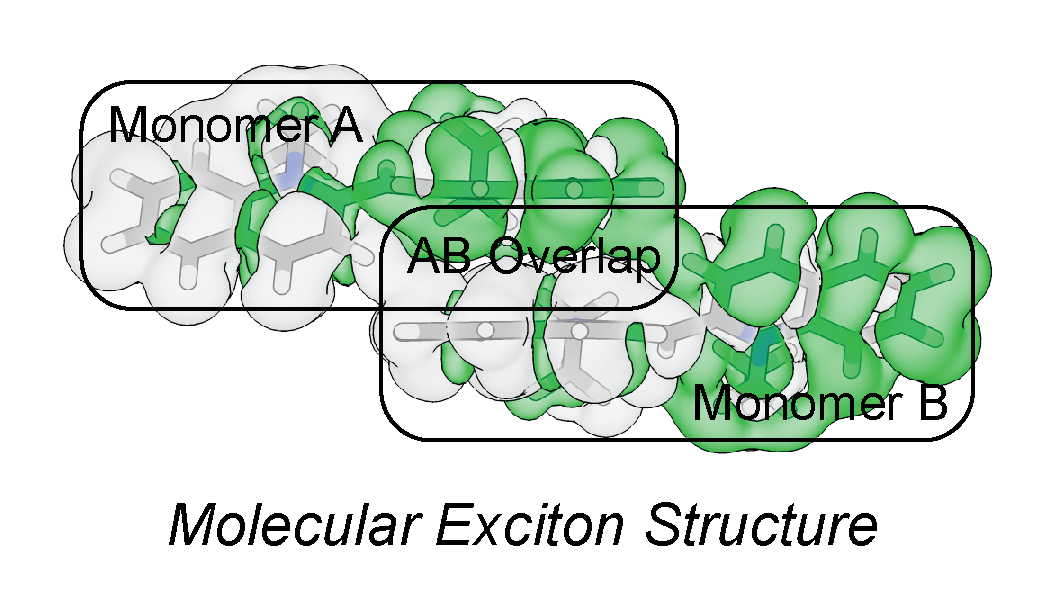
When chromophores aggregate, the electronic structure of an excited state can become delocalized over two or more molecular units creating an exciton. The relative orientation and magnitude of the molecule’s electronic transition dipoles will dictate the spectral and energy transfer properties of the exciton. By modifying the molecular aggregate structure, unique electronic and energy transfer properties can be designed. We aim to develop a computational toolset to calculate electronic interactions of molecular aggregates, and to predict their excitonic functionality, which could be applied towards building synthetic biological energy transfer systems using DNA origami as a scaffold.
Electronic and molecular modeling
Modern computational chemistry methods, including electronic structure theory and all-atom molecular dynamics, can be used to probe the unique properties of biomolecular systems at length- and time-scales inaccessible to experimental methods. In addition, the recent application of GPU hardware to dramatically speed up biomolecular simulation has expanded the size and complexity of systems that can be modeled. We utilize existing computational chemistry software as well as develop our own modeling tools using modern scripting languages such as Python.
Funding Sources
Organization of chromophore assemblies
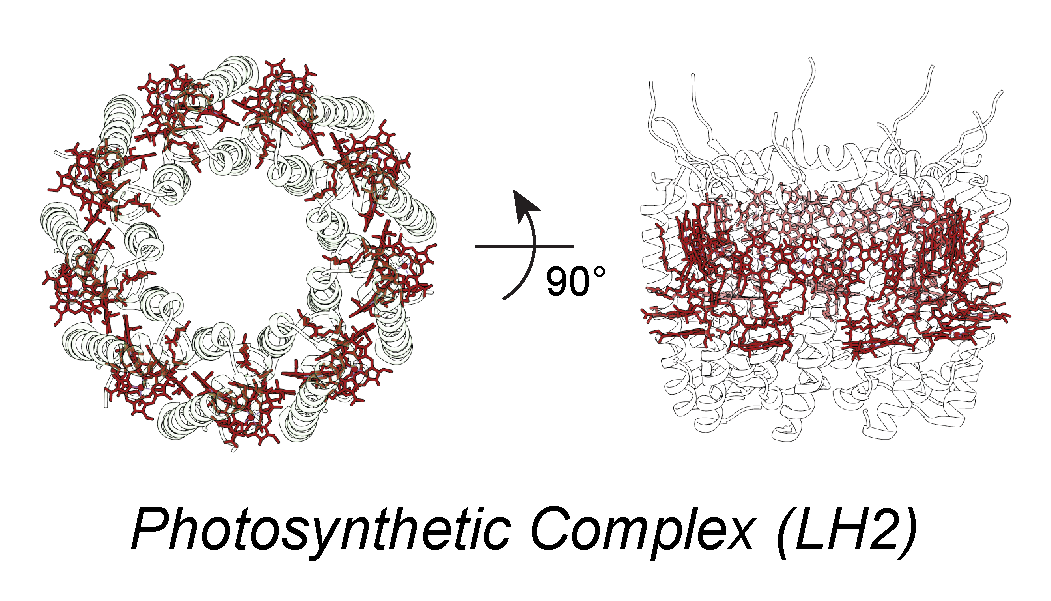
Biomolecular light-harvesting complexes in nature utilize a combination of short- and long-range energy transfer mechanisms to efficiently harvest sunlight and to avoid energy dissipation. In addition, different species of chromophores are present within the same complexes, yielding varying spectral properties in order to direct energy transfer via a downhill energetics funnel. With the inclusion of the relative orientation of molecules, the possible parameter space for designing biosynthetic energy transport systems is essentially intractable. We aim to reduce the parameter space by applying a systems-level optimization of chromophore organization to determine the relative importance of each parameter.
Expanding the junction space for DNA nanotechnology
The DNA four-way junction (4WJ) or Holliday junction is used as a primary building block in DNA origami, where two strands can cross from one duplex to another to “staple” the duplexes together. This double-crossover motif, when repeated sequentially, can be used to build arbitrary 2D and 3D wireframe DNA structures. The 4WJ structure has been shown to be sensitive to the core junction sequence, leading to distinct isomerization states (Adendorff, Nucleic Acids Res., 2022), which can affect the stability of DNA origami molecules built out of this junction motif. We are currently investigating novel junction motifs for DNA nanotechnology using all-atom molecular dynamics, including heterochiral DNA junctions including both right- and left-handed DNA regions.